keumuman
Alport syndrome adalah kelainan genetik herediter yang jarang yang menyebabkan hilangnya fungsi ginjal, gangguan pendengaran dan anomali mata secara progresif.

Penyebab sindrom Alport adalah mutasi gen yang terlibat dalam produksi protein penting untuk berfungsinya ginjal, telinga bagian dalam dan mata.
Diagnosis sindrom Alport didasarkan pada pemeriksaan fisik, riwayat medis, biopsi ginjal, dan tes genetik.
Saat ini, mereka yang menderita sindrom Alport hanya dapat mengandalkan perawatan simtomatik, yaitu mereka mengurangi gejala dan menunda komplikasi.
Apa itu Sindrom Alport?
Alport syndrome adalah kelainan genetik langka yang menyebabkan masalah ginjal yang parah, gangguan pendengaran dan kelainan okular pada mereka yang membawanya.
Alport syndrome adalah suatu kondisi yang diturunkan, di mana istilah "herediter" berarti "ditularkan oleh satu atau kedua orang tua".
Epidemiologi: seberapa umum sindrom Alport?
Menurut statistik, satu orang setiap 50.000 akan dilahirkan dengan sindrom Alport.
sinonim
Karena sifat bawaannya dan karena keterlibatan ginjalnya, sindrom Alport juga dikenal, dalam bidang medis, sebagai nefritis herediter .
penyebab
Sindrom Alport disebabkan oleh mutasi - yang merupakan perubahan abnormal - dalam satu atau lebih dari 3 gen manusia yang dinamai dengan huruf COL4A3, COL4A4 dan COL4A5 .
Di mana gen yang bertanggung jawab atas sindrom Alport berada?
Sedangkan untuk gen COL4A3 dan COL4A4 lokalisasi adalah untuk kedua kromosom 2 dari genom manusia, untuk gen COL4A5 itu pada kromosom X dari genom manusia.
Kromosom 2 adalah kromosom autosom ; kromosom X, di sisi lain, adalah kromosom seks, yang merupakan kromosom di mana jenis kelamin individu bergantung.
Efek mutasi
Premis: gen yang ada pada kromosom manusia adalah sekuens DNA yang memiliki tugas menghasilkan protein mendasar dalam proses biologis yang sangat diperlukan untuk kehidupan, termasuk pertumbuhan dan replikasi sel.
Ketika mereka bebas dari mutasi (oleh karena itu pada orang yang sehat), 3 gen yang terkait dengan sindrom Alport menghasilkan komponen protein mendasar untuk generasi yang benar, pemrosesan dan penataan akhir dari kolagen tipe IV yang lazim di ginjal, di telinga bagian dalam dan di mata .
Ketika sebaliknya mereka menjadi korban mutasi, 3 gen yang terkait dengan sindrom Alport kehilangan kemampuan untuk menghasilkan komponen protein yang sangat diperlukan untuk realisasi kolagen tipe IV yang disebutkan di atas dan ini melibatkan perubahan jaringan pada ginjal, telinga bagian dalam dan mata, dengan disfungsi akibat organ-organ ini.
Tahukah Anda bahwa ...
Keterlibatan kolagen tipe IV, oleh sindrom Alport, membuatnya menjadi penyakit jaringan ikat, seperti sindrom Ehlers Danlos atau sindrom Marfan .
FISIOPATOLGIA: BEBERAPA DETAIL TAMBAHAN
Gen yang terkait dengan sindrom Alport berkontribusi pada pembentukan varian kolagen tipe IV, yang penting untuk:
- Tindakan penyaringan yang benar dari glomeruli ginjal terhadap darah.
Penjelasan. Terletak di ginjal, glomeruli ginjal adalah aglomerat pembuluh darah tertentu, yang mampu menghilangkan (seolah-olah mereka menyaring) zat-zat limbah dari darah dan membuat urin.
Dengan perubahan kolagen tipe IV mereka, struktur ginjal ini gagal dalam tindakan penyaringan dan pembuatan urin.

- Karya terjemahan, dibuat oleh organ Corti dari telinga bagian dalam, dari gelombang suara di impuls saraf.
Penjelasan. Organ Corti adalah struktur telinga bagian dalam yang bertanggung jawab untuk mengubah gelombang suara yang dirasakan oleh telinga menjadi sinyal saraf "yang dapat dibaca" dan "dapat ditafsirkan" dari otak manusia.
Perubahan kolagen tipe IV yang dikombinasikan dengan organ Corti merusak proses penerjemahan suara ke dalam bahasa sistem saraf manusia.
- Mempertahankan bentuk lensa yang tepat, kekakuan kornea yang memadai, dan warna normal dari epitel pigmen retina.
Penjelasan. Bentuk lensa kristal dan kekakuan kornea sangat diperlukan untuk fungsi visual dan kesehatan mata (warna epitel pigmen retina, di sisi lain, tampaknya tidak memiliki kepentingan di area ini).
Kehadiran di mata kolagen tipe IV yang diubah memodifikasi bentuk lensa kristal, kekakuan kornea dan warna epitel pigmen retina.
Semua informasi ini menjelaskan mengapa sindrom Alport dikaitkan dengan gejala tertentu.
Warisan sindrom Alport
Untuk memahami ...
- Setiap gen manusia hadir dalam dua salinan, yang disebut alel, satu dari ibu dan satu dari ayah.
- Penyakit bawaan adalah autosom dominan ketika, terjadi, mutasi hanya satu salinan gen yang menyebabkannya cukup.
- Penyakit yang diturunkan adalah resesif autosomal ketika mutasi kedua salinan gen yang menyebabkannya diperlukan untuk terjadi.
Alport syndrome menyajikan 3 model pewarisan yang berbeda .
Menurut pola pewarisan yang paling umum (80% kasus), sindrom Alport adalah penyakit bawaan yang dikaitkan dengan kromosom X (seperti hemofilia atau kebutaan warna), karena tergantung pada mutasi gen COL4A5 yang terletak pada kromosom. X seksual
Menurut model pewarisan paling umum kedua (sekitar 10% dari kasus), sindrom Alport berperilaku seperti apa yang disebut penyakit bawaan resesif autosom, karena karena keberadaannya, mutasi spesifik diperlukan di kedua alel salah satu gen COL4A3 dan COL4A4, terletak di kromosom 2.
Akhirnya, sesuai dengan model pewarisan yang kurang umum (sekitar 5% kasus), sindrom Alport mewakili contoh penyakit bawaan dominan autosom, karena mutasi spesifik pada salah satu alel gen COL4A3 dan COL4A4 cukup untuk manifestasinya. .
ALPORT SYNDROME TERKAIT DENGAN CHROMOSOME X
Sindrom Alport X-linked memiliki efek terburuk pada subjek pria.
Fenomena ini terkait dengan set kromosom seksual yang menjadi ciri pria dan wanita; pada wanita, pada kenyataannya, ada dua kromosom X, yang, dengan adanya mutasi terhadap satu dari dua, saling membantu (yang sehat mengkompensasi, lebih atau kurang efektif, untuk kesalahan yang diubah); pada manusia, bagaimanapun, hanya ada satu kromosom X (yang lainnya adalah kromosom seks Y), yang, jika mengalami mutasi, tidak dapat mengandalkan dukungan dari kromosom lain dengan fungsi yang sama.
SINDROM ALPOR RESESIF OTOSOMI
Alport syndrome dengan perilaku resesif autosom menyebabkan gejala dan tanda-tanda serupa pada pria dan wanita.
SINDROM ALPOR OTOMATIS DOMINAN
Sindrom Alport dengan perilaku dominan autosom memengaruhi pria dan wanita dengan tingkat keparahan yang sama.
Gejala dan Komplikasi
Karena kurangnya kolagen tipe IV yang memadai di ginjal, telinga bagian dalam dan mata, sindrom Alport secara klasik menentukan:
- Hilangnya fungsi ginjal yang progresif dan tak terhindarkan,
- Penurunan kemampuan pendengaran e
- Kelainan mata dikombinasikan dengan defisit visual .
Dalam beberapa kasus yang jarang, sindrom Alport memiliki efek yang luas sehingga, dengan konsekuensi yang ditunjukkan di atas, ia juga menambahkan leiomiomatosis luas pada pohon esofagus dan trakeobronkial, dan masalah vaskular yang serius ( diseksi aorta dan aneurisma aorta toraks atau abdomen atau toraks aorta ).
Kehilangan fungsi ginjal: gejala
Premis: dengan ungkapan "hilangnya fungsi ginjal", dokter bermaksud bahwa ginjal dan struktur internalnya (misalnya glomeruli ginjal) mengalami penurunan bertahap dalam kapasitas penyaringan dan produksi urin.
Di hadapan sindrom Alport, gejala yang berkaitan dengan hilangnya fungsi ginjal terutama terdiri dari hematuria, yaitu darah dalam urin, dan proteinuria, yaitu adanya protein dalam urin.
Hematuria adalah gejala yang cukup dini, dalam arti bahwa, pada subjek dengan sindrom Alport, ia cenderung muncul pada usia muda; dalam banyak kasus, pengenalannya hanya dimungkinkan melalui mikroskop ( hematuria mikroskopis ).
Sebaliknya, proteinuria adalah gejala kemudian, yaitu, yang muncul ketika sindrom Alport berada pada tahap yang lebih lanjut.
Kemampuan mendengar menurun: detail
Pada tingkat pendengaran, sindrom Alport menyebabkan ketulian sebagian ; tepatnya, itu menginduksi gangguan pendengaran sensorineural, yang mencegah pasien dari mendengar suara pada frekuensi tinggi ( gangguan pendengaran frekuensi tinggi).
Perkembangan masalah pendengaran pada orang-orang yang menderita sindrom Alport bervariasi dalam kaitannya dengan subjek gen yang mengalami mutasi dan jenis pewarisan; Bahkan:
- Jika mutasi berada di COL4A5, laki-laki mengalami gangguan pendengaran menjelang akhir masa kanak-kanak, sementara perempuan, dalam keadaan langka yang mereka minati, kembangkan di usia tua;
- Jika mutasi terjadi pada COL4A4 atau COL4A3 dan penyakitnya adalah tipe resesif autosom, baik pasien pria maupun pasien wanita mengeluh tentang gejala pertama gangguan pendengaran pada akhir masa kanak-kanak atau remaja awal;
- Jika mutasi tinggal di COL4A4 atau COL4A3 dan penyakitnya dominan autosom, pasien pria dan wanita mengalami gangguan pendengaran sensorineural di usia tua.

Untuk alasan yang masih belum diketahui, sindrom Alport menyelamatkan beberapa pasien dari masalah pendengaran; dengan kata lain, untuk alasan yang tidak diketahui, beberapa pembawa sindrom Alport tidak mengalami gangguan pendengaran sensorineural.
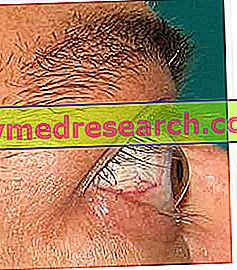
Anomali mata dan defisit visual: detail
Hanya dapat diamati pada persentase tertentu dari pasien, anomali okular yang disebabkan oleh sindrom Alport disebabkan oleh kondisi seperti: keratoconus, lenticonous, katarak dan adanya bintik - bintik pada makula retina .
Kecuali untuk adanya bintik-bintik pada makula retina, semua kondisi ini hanya mempengaruhi kurang lebih kemampuan visual pasien.
Leiomiomatosis difus pada pohon esofagus dan trakeobronkial
Leiomiomatosis difus kerongkongan dan pohon trakeobronkial adalah tumor jinak langka yang, pada subjek dengan sindrom Alport, dapat menyebabkan:
- disfagia;
- Muntah postprandial;
- Nyeri epigastrium dan nyeri retrosternal;
- Bronkitis berulang;
- dyspnea;
- batuk;
- Melengking saat bernafas.
komplikasi
Komplikasi utama sindrom Alport adalah keadaan gagal ginjal stadium akhir, akibat hilangnya fungsi ginjal yang progresif dan tak terhindarkan.
Dalam kedokteran, istilah "gagal ginjal tahap akhir" menunjukkan tahap akhir dan juga tahap paling serius dari gagal ginjal kronis, yaitu kondisi di mana ginjal telah kehilangan semua kapasitas fungsionalnya secara lengkap dan definitif.
Bertanggung jawab untuk konsekuensi yang sangat serius (misalnya: hipertensi, edema paru, kerapuhan tulang, imunosupresi, kerusakan sistem saraf, dll.), Gagal ginjal stadium akhir membutuhkan perawatan kronis seperti dialisis dan merupakan indikasi terpenting untuk operasi. bedah seperti transplantasi ginjal .
KOMPLIKASI LAINNYA
Kemungkinan komplikasi lain dari sindrom Alport, yang juga memiliki tingkat kematian yang tinggi, adalah pendarahan internal yang mengikuti laserasi diseksi aorta atau akhirnya aneurisma aorta toraks atau abdominal.
diagnosa
Sebagai aturan, untuk diagnosis sindrom Alport, dokter menggunakan informasi dari:
- Pemeriksaan fisik.
- The anamnesis ;
- Biopsi ginjal ;
- Tes genetik .
Pemeriksaan fisik
Ini adalah pengamatan medis dari gejala dan tanda yang ditunjukkan oleh pasien.
Dalam konteks sindrom Alport, itu mengungkapkan bagian dari masalah ginjal, masalah pendengaran (jika ada) dan anomali okular (jika ada).
sejarah
Ini adalah studi kritis terhadap simptomatologi melalui pertanyaan spesifik, dikombinasikan dengan pemeriksaan riwayat keluarga pasien.
Sebagai bagian dari prosedur pemeriksaan yang berfungsi untuk mengenali sindrom Alport, anamnesis memungkinkan dokter untuk mengetahui saat yang tepat dari timbulnya gejala dan untuk mengetahui apakah, dalam keluarga pasien, beberapa penyakit keturunan terjadi.
Biopsi ginjal
Terdiri dari pengumpulan dan analisis laboratorium dari sampel sel-sel ginjal.
Dalam konteks sindrom Alport, berguna untuk menilai keadaan kesehatan ginjal dan untuk memastikan tidak adanya kolagen tipe IV fungsional.
Biopsi ginjal adalah langkah penting dalam diagnosis sindrom Alport, karena, jika tepat waktu, terapi yang paling tepat dapat direncanakan sesegera mungkin.
Tes genetika
Ini adalah analisis DNA yang bertujuan mendeteksi mutasi gen kritis.
Dalam konteks sindrom Alport, ini mewakili tes konfirmasi diagnostik dan pemeriksaan yang digunakan untuk menentukan gen mutasi yang tepat dan jenis pewarisan.
terapi
Dari sindrom Alport tidak mungkin disembuhkan ; Namun, mereka yang terkena dampak dapat mengandalkan berbagai perawatan simptomatik, yang memungkinkan untuk mengontrol gejala dan menunda timbulnya komplikasi.
Tahukah Anda bahwa ...
Untuk dapat pulih dari penyakit seperti sindrom Alport, perlu untuk menghilangkan mutasi genetik atau membatalkan efeknya sedemikian rupa sehingga kolagen tipe IV normal dan fungsional.
Terapi simtomatik: terdiri dari apakah itu?
Daftar perawatan simtomatik untuk pengelolaan sindrom Alport meliputi:
- Terapi farmakologis berdasarkan inhibitor ACE ;
- Terapi farmakologis klasik dipertimbangkan dengan adanya gagal ginjal kronis;
- Dialisis;
- Transplantasi ginjal;
- Penggunaan alat bantu dengar ;
- Perawatan untuk keratoconus, lenticonous dan / atau katarak.
INHIBITOR ACE
Biasanya diindikasikan untuk pengobatan hipertensi dan beberapa penyakit kardiovaskular, ACE inhibitor digunakan dengan adanya sindrom Alport, berdasarkan kemampuan mereka yang diperlihatkan untuk memperlambat penurunan fungsi ginjal secara progresif, tipikal dari penyakit yang diturunkan yang disebutkan sebelumnya.
Berkat kemampuan ini, oleh karena itu, ACE inhibitor memungkinkan penundaan gagal ginjal dan perlunya perawatan penting seperti dialisis atau transplantasi ginjal.
Untuk alasan yang tidak diketahui, pada beberapa individu dengan sindrom Alport, ACE inhibitor tidak efektif; ini menjelaskan mengapa dokter mencari obat dengan kekuatan yang sama.
TERAPI FARMAKOLOGIS UNTUK KEGAGALAN Renovasi KRONIS
Ketika sindrom Alport mengakibatkan gagal ginjal kronis, ada beberapa obat yang bermanfaat, termasuk:
- Obat-obatan melawan hipertensi (di antaranya, termasuk penghambat ACE dan ARB yang telah disebutkan, dan diuretik);
- Suplemen kalsium dan vitamin D (untuk melindungi tulang dari patah tulang);
- Sodium polystyrene sulfonate dan analog (untuk mencegah akumulasi kalium dalam darah).
DIALISIS DAN TRANSPLANTASI GINJAL
Dialisis adalah perawatan yang secara artifisial mereproduksi fungsi-fungsi tertentu dari ginjal, membersihkan darah dari sisa produk dan air.
Transplantasi ginjal, di sisi lain, adalah operasi untuk menggantikan satu atau kedua ginjal dengan ginjal yang sehat, dari donor yang kompatibel (donor mungkin tidak hidup atau hidup).
Baik dialisis dan transplantasi ginjal diindikasikan ketika sindrom Alport berada pada tahap paling lanjut, yaitu ketika ada gagal ginjal stadium akhir.
APARATUS AKUSTIK
Di hadapan sindrom Alport, alat bantu dengar adalah obat yang diindikasikan untuk semua pasien dengan tuli parsial.
PENGOBATAN KERATOCON, LENTICON DAN CATARACT
Ketika sindrom Alport memengaruhi mata, menyebabkan keratoconus, lenticonous dan / atau katarak, pasien dapat mengandalkan operasi mata, yang hasilnya lebih dari memuaskan.
Tokoh medis apa yang dilibatkan terapi simptomatik sindrom Alport?
Pengobatan simtomatik sindrom Alport memerlukan intervensi terkoordinasi dari beberapa spesialis medis, termasuk: dokter anak, ahli nefrologi, audiolog dan dokter mata.
prognosa
Alport syndrome memiliki prognosis yang berbeda tergantung pada gen yang terkait dengan keberadaannya; Namun, secara umum, itu tidak pernah baik, karena, kondisi yang tidak dapat disembuhkan terpisah, setiap pasien ditakdirkan untuk menderita kehilangan fungsi ginjal.
pencegahan
Alport syndrome adalah kondisi yang tidak mungkin untuk dicegah .